Génétique, épigénétique et biologie des sarcomes

Objectifs
Les sarcomes forment un ensemble hétérogène de tumeurs malignes agressives d’origines mésenchymateuse. Ils touchent environ 4000 personnes par an en France et sont plutôt de mauvais pronostic. Malgré leur relative faible nombre chez l’adulte, les sarcomes sont responsables de plus de 20% de la mortalité liée au cancer chez les enfants et les jeunes adultes.
En raison de leur grande hétérogénéité (plus de 50 types histologiques différents et plus de 150 sous-types moléculaires) le diagnostic précis des sarcomes est souvent difficile à poser, générant une prise en charge thérapeutique difficile. Grâce au séquençage à haut débit, l’identification de nouvelles altérations a considérablement amélioré le diagnostic et a fourni des indications sur les mécanismes oncogéniques.
Notre équipe a pour objectifs :
- De permettre une caractérisation moléculaire des sarcomes la plus exhaustive possible,
- D’étudier fonctionnellement les altérations identifiées dans l’objectif 1 permettant de découvrir de nouveaux mécanismes oncogéniques,
- D’identifier des gènes, des modifications épigénétiques ou des voies de signalisation nécessaires à la croissance tumorale et permettant le développement d’approches thérapeutiques ciblées innovantes.
![]()
Projets
Classification moléculaire des sarcomes
En étroite collaboration avec le département de Pathologie du Centre Léon Bérard nous avons entrepris une classification moléculaire des sarcomes en utilisant le séquençage ARN à haut débit à partir d’échantillon tumoraux préservés en bloc de paraffine.
A partir de ce séquençage nous pouvons déterminer pour chaque échantillon la présence de gènes de fusions, le profil d’expression –dont les profils immunitaires spécifiques- et les variants nucléotidiques codants, que nous mettons en relation avec les données cliniques et d’anatomopathologie. Notamment, l’accumulation de plusieurs milliers de profils d’expression tumoraux, nous permet, par des méthodes non supervisées et/ou d’apprentissage, de classifier les échantillons tumoraux (Figure 1).
Cette stratégie nous a déjà permis de mettre en évidence de nouveaux sous-types tumoraux, comme les sarcomes thoraciques SMARCA4-déficient (Le Loarer F. et al, 2014, Perret, R et al. 2018), les rhabdomyosarcomes à fusion EWSR1/FUS-TFCP2 (Watson et al. 2018, Le Loarer F. et al, 2019), ou encore les sarcomes à petites cellules à translocation CRTC1-SS18 (Alholle et al 2019). Ces analyses sont également menées pour d’autres pathologies cancéreuses comme les tumeurs mélaniques (en collaboration avec le Dr Arnaud de la Fouchardière, CLB) et les mésothéliomes (Dr Françoise Galateau-Sallé, CLB).
Enfin, au sein de notre projet iMAPS (INCa-DGOS_13219), nous engageons une analyse intégrative des sarcomes pédiatriques inclassés en étudiant non seulement leur expression génique mais également leur méthylome et exome.

Figure 1 : analyse non supervisée (t-SNE) des profils d’expression tumoraux montrant le regroupement des échantillons (symbolisées par des sphères) par sous-types tumoraux (représentés par des couleurs différentes).
Etudes fonctionnelles des protéines de fusions impliquées dans les sarcomes
Ces dernières années, de nombreux nouveaux gènes de fusion, oncogènes responsables du développement cancéreux, ont été identifiés, en particulier dans les sarcomes. Parmi ceux-ci, nous nous intéressons à ceux observés le plus fréquemment et/ou qui ne sont pas ou peu étudiés. Grâce à la caractérisation moléculaire décrite ci-dessus, nous avons identifié quelques gènes de fusion récurrents non encore décrits que nous explorons fonctionnellement.
En particulier, nous nous sommes focalisés sur l’étude fonctionnelle des gènes de fusion CIC-DUX4 et HEY1-NCOA2 et du gène BCOR réarrangé, qui n’ont bénéficié au mieux que d’études fonctionnelle très succinctes, dans des modèles cellulaires ectopiques. Grâce au développement de modèles cellulaires spécifiques (en collaboration avec les équipes d’Olivier Delattre, Katia Scotlandi et James Amatruda) nous sommes en mesure d’étudier ces gènes aberrants dans leur contexte naturel.
En les inhibant dans des modèles cellulaires pertinents, nous pouvons déterminer, lors d’expériences de ChIP-seq, d’immunoprécipitations suivies d’une analyse en spectrométrie de masse et de RNA-seq, les cibles directes de ces oncoprotéines ainsi que leurs partenaires protéiques. Nous étudions également les voies de signalisation dérégulées dans ces tumeurs afin d’identifier des cibles thérapeutiques potentielles que nous pouvons ensuite cibler dans nos modèles in vivo.
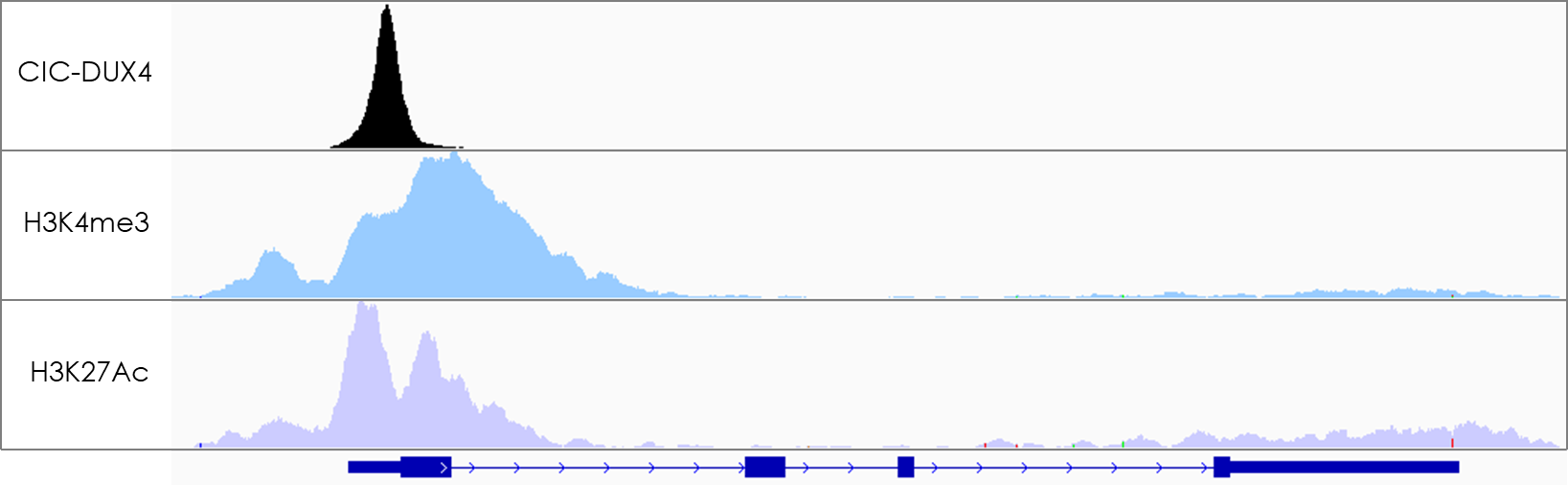
Figure 2 : ChIP-seq montrant un site de fixation de CIC-DUX4 dans une région promotrice (positive pour les marques activatrices H3K4me3 et H3K27Ac).
Rôle de l’épigénétique dans l’oncogenèse des sarcomes
L’initiation et la progression du cancer sont contrôlées par des événements aussi bien génétiques qu’épigénétiques. Contrairement aux modifications génétiques, les aberrations épigénétiques sont réversibles, ce qui permet théoriquement de pouvoir rétablir l’état épigénétique des cellules malignes à leur état pré-malin.
Par ailleurs, nous avons pu établir que nombres de sarcomes à translocation possèdent un profil transcriptomique évoquant un défaut dans le processus de remodelage de la chromatine ou plus généralement dans les mécanismes épigénétiques.
Nous étudions particulièrement les mécanismes épigénétiques dérégulés dans les sarcomes BCOR-réarrangés et dans les sarcomes du stroma de l’endomètre possédant une altération de BCOR (ou d’un gène assimilé), BCOR étant un membre du complexe répressif PRC1.1 du remodelage de la chromatine.
-
Franck Tirode,
DR2 INSERM
Chef d’équipe
franck.tirode@lyon.unicancer.frSuivez nous aussi sur :
Nous recrutons! Voir l’annonce ici.